The Turkish Journal of Pediatrics 2007; 49: 444-447Axenfeld-Rieger syndrome associated with truncus arteriosus: a case report
Özlem Gürbüz-Köz1, Tuba Atalay1, Cem Köz2, Hatice Ilgın Ruhi3Alper Yarangümeli1, Gülcan Kural111st Eye Clinic, Ankara Numune Training and Research Hospital, 2Department of Cardiology, Gülhane Military Academy of Medicine, and 3Department of Medical Genetics, Ankara University Faculty of Medicine, Ankara, TurkeySUMMARY: Gürbüz-Köz Ö, Atalay T, Köz C, Ilgın-Ruhi H, Yarangümeli A, Kural G. Axenfeld-Rieger syndrome associated with truncus arteriosus: a case report. Turk J Pediatr 2007; 49: 444-447. The aim of this presentation was to report a case with Axenfeld-Rieger syndrome (ARS) associated with truncus arteriosus (TA). We present a 14-year-old boy with ARS in whom the diagnosis was confirmed by ophthalmologic examination and developmental defects of the teeth and facial bones. Echocardiography revealed TA. With this case demonstrating the association between ARS and TA, the range of reported cardiac malformations is enlarged and the importance of cardiologic evaluation is emphasized in patients with ARS. Key words: Axenfeld-Rieger syndrome, truncus arteriosus.
Axenfeld-Rieger syndrome (ARS) is a clinically
In ARS, classical signs represented by dental
and genetically heterogeneous disorder with
hypoplasia, craniofacial anomalies, and involuted
an autosomal dominant mode of transmission
periumbilical skin can be associated with a wide
and great intrafamilial variability, consisting of
diversity of other traits, such as limb anomalies,
a family of developmental diseases including
short stature, pituitary anomalies, empty sella
anterior segment abnormalities and a variety
syndrome, and a variety of neurological and
Axenfeld-Rieger syndrome can be classified
Among the associated extraocular features,
as Axenfeld anomaly (limited to peripheral
cardiac malformations, including interatrial
anterior segment defects), Rieger anomaly
septal defects and semilunar valve stenosis or
(peripheral abnormalities with additional
insufficiencies, have rarely been reported5-9.
changes in the iris), and Rieger syndrome
Here we present a case of ARS with truncus
(ocular anomalies and extraocular developmental
arteriosus (TA) type IV. This is the first case
defects especially of the teeth, facial bones, and
periumbilical skin). Because of the marked genotypic and phenotypic overlap, it has been
Case Report
proposed that these diseases are best considered
The patient was a 14-year-old boy born from
under the single ARS heading. These three
nonconsanguineous healthy parents. He was the
variations are now recognized as a spectrum of
9th sibling of the family. The pregnancy was
the same syndrome1-3. In the literature, cases
normal without any exposure to teratogenic
with ocular and extraocular manifestations are
agents. The delivery was also normal at term.
either defined as ARS or Rieger syndrome4.
The family history did not reveal any other
The most important ocular feature of the ARS
is glaucoma, which develops in about 50%
The patient had been referred to our clinic
of affected individuals. The ocular anomalies
because of bilateral gradual visual impairment.
are suggested to represent an arrest of tissues
Best corrected visual acuity was 0.3 in the
derived from neural crest cells in gestation1.
right eye and 0.1 in the left eye. Slit lamp
Axenfeld-Rieger Syndrome with Truncus Arteriosus 445
examination displayed a prominent, anteriorly
latanoprost 0.005% (Xalatan, Pharmacia &
displaced Schwalbe’s line in all quadrants of
Upjohn, Uppsala, Sweden) once daily at 10:00
both eyes. The iris had stromal hypoplasia
pm, and after one month of therapy, intraocular
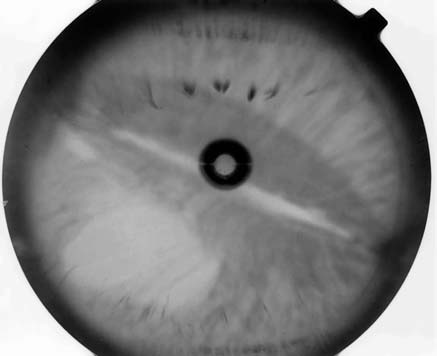
bilaterally. There were corectopia in the right
pressure was 18 mmHg in the left eye.
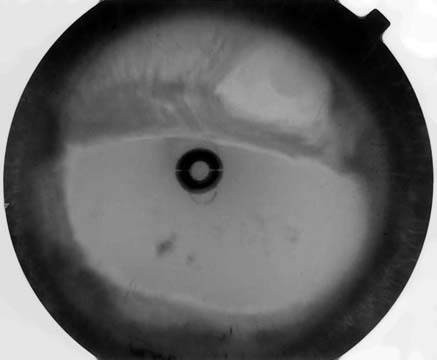
and polycoria in the left eye (Figs. 1 and 2).
Non-ocular abnormalities consisted of facial
Gonioscopy revealed iris strands attached to
configuration with flattening of the mid-face, a
the Schwalbe line in both eyes. Cup/disc ratios
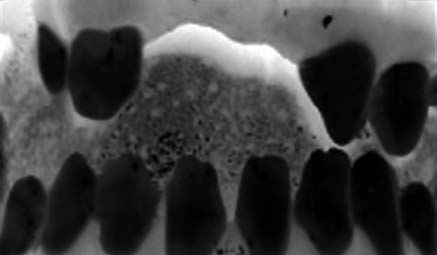
thin upper lip, and protruding lower lip (Fig. 3).
were 0.3 in both eyes. Intraocular pressures
Hypodontia and microdontia were also present
(Fig. 4). Physical examination revealed the
in the left eye. Results of visual field testing
absence of redundant skin around the umbilicus.
with automated perimetry were fairly reliable
We considered this patient’s ocular and non-
in both eyes. Nonspecific visual field defects
ocular abnormalities to be typical of ARS. After
were detected because of pupillary distortion
informed consent was obtained, peripheral blood
in both eyes. There were no anomalies at the
sample was obtained. Chromosome analysis
lens or fundus. The patient was given topical
revealed a 46-XY normal male karyotype. Fig. 1. Biomicroscopic appearance of the right eye Fig. 2. Biomicroscopic appearance of the left eye
showing distortion and displacement of pupil with
showing distortion and displacement of pupil with full-
thinning of iridic stroma, peripheric anterior synechiae
thickness hole formation and thinning of iridic stroma. Fig. 3. The patient’s facial configuration with flattening
of the mid-face, thin upper lip, protruding
Fig. 4. Photograph illustrating dental anomalies such as The Turkish Journal of Pediatrics • October - December 2007
Owing to clubbing and history of shortness
ARS and TA, the range of reported cardiac
of breath, the patient was referred to a
malformations is enlarged and the importance
cardiologist, who found a soft systolic thrill
of cardiologic evaluation is emphasized in
along the left sternal border and loud apical
pansystolic murmur at the left lower sternal
border, radiating to the whole precordial
accompanying ARS have been described. In
area and especially to the right side. A two-
1994, Tsai et al.5 described a patient with aortic
dimensional echocardiographic examination
stenosis associated with ARS. Cunningham
revealed TA (Figs. 5 and 6). The patient was
et al.6 then described ARS coexisting with
admitted to another hospital for cardiac surgical
atrial septal defects and sensorineural hearing
loss, affecting multiple members of a single family. In 2000 Bekir et al.7 reported a 20-year-old girl with ARS associated with an atrial septal defect and interatrial aneurysm. Recently, Baruch and Erickson8 described two siblings presenting with ARS, hypertelorism, clinodactyly, and cardiac anomalies such as patent ductus arteriosus and atrial septal defect. Most recently, Grosso et al.9 reported a family with a clinical picture overlapping that described by Cunningham et al.6 and characterized by ARS in association with cardiac malformations and sensorineural deafness, without facial dysmorphisms, dental hypoplasia, or involuted periumbilical skin. However, in their patients,
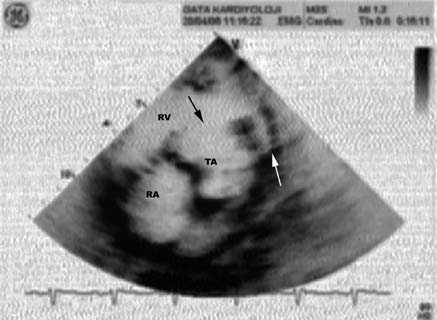
Fig. 5. Echocardiographic characterization of truncus
cardiac malformations were represented by
arteriosus is ventricular septal defect (arrow) with
truncus arteriosus. LV: Left ventricle. RV: Right
mitral and tricuspid valve defects instead of the
atrial septal defects observed in the patients of Cunningham et al.6.
Many hypotheses have been proposed for the pathogenesis of ARS on the assumption that the lesions have a common developmental origin during embryonic life. Because subsequent research has shown that the involved ocular tissues originate from the neural crest, ARS is now theorized as developing from the abnormal migration of neural crest cells10,11, despite this disease having first been described as mesodermal dysgenesis. Shields11 has proposed that there is a developmental arrest of certain anterior segment structures derived from neural crest cells that leave primordial endothelial layer on portions of the iris and anterior chamber
Fig. 6. Truncus arteriosus type IV, ventricular septal
angle, appearing to account for the iridocorneal
defect (white arrow) with pulmonary arterial agenesis
(black arrow). LV: Left ventricle. RV: Right ventricle.
strands and the changes in the central iris.
A developmental arrest of neural crest tissue is believed to account for the ocular and
Discussion
most of the systemic abnormalities in ARS1-3.
Embryological studies demonstrated that neural
malformations has been described, but TA
crest cells play a key role in the development
has not been reported before5-9. With this
of cardiac structures such as the outflow
case demonstrating the association between
tract and the aortic arch system. Takamura
Axenfeld-Rieger Syndrome with Truncus Arteriosus 447
and associates12 have also shown that neural
4. Perveen R, Lloyd IC, Clayton-Smith J, et al. Phenotypic
crest cells are intimately associated with the
variability and asymmetry of Rieger syndrome associated with PITX2 mutations. Invest Ophthalmol
formation of both the aortic and pulmonic
5. Tsai JC, Grajewski AL. Cardiac valvular disease and
Axenfeld-Rieger syndrome. Am J Ophthalmol 1994;
documented with association to chromosome
413 chromosome 614, and chromosome 1315.
6. Cunningham ET Jr, Eliott D, Miller NR, Maumenee
IH, Green WR. Familial Axenfeld-Rieger anomaly,
with association to these chromosomes16-18.
atrial septal defect, and sensorineural hearing loss: a possible new genetic syndrome. Arch Ophthalmol
Conventional cytogenetic analysis in our case
revealed a normal male karyotype 46-XY, but
7. Bekir NA, Güngör K. Atrial septal defect with
further evaluation is crucial to determine the
interatrial aneurysm and Axenfeld-Rieger syndrome.
genetic role in ARS and TA association.
Acta Ophthalmol Scand 2000; 78: 101-103.
In the families described by Grosso et al.9 and
8. Baruch AC, Erickson RP. Axenfeld-Rieger anomaly,
Cunningham et al.6, ARS, cardiac malformations
hypertelorism, clinodactyly, and cardiac anomalies in
and sensorineural hearing loss were present.
sibs with an unbalanced translocation der(6)t(6;8). Am J Med Genet 2001; 100: 187-190.
The difference between these families in terms of cardiac anomalies was interpreted as the
9. Gross S, Farnetani MA, Berardi R, et al. Familial
Axenfeld-Rieger anomaly, cardiac malformations, and
variable expression of the same genetic defect.
sensorineural hearing loss: a provisionally unique genetic
Grosso et al.9 indicated that the inherited
syndrome? Am J Med Genet 2002; 111: 182-186.
traits in members of the presented families
10. Bahn CF, Falls HF, Varley GA, Meyer RF, Edelhauser
were connected and were not coincidental
HF, Baurne WM. Classification of corneal endothelial
and proposed that patients were affected by
disorders based on neural crest origin. Ophthalmology
a provisionally unique genetic syndrome as
hypothesized by Cunningham et al.6. Genetic
11. Shields MB, Buckley E, Klintworth GK, Thresher R.
studies will clarify whether they manifest
Axenfeld-Rieger syndrome. A spectrum of developmental
a unique phenotypic expression of ARS or
disorders. Surv Ophthalmol 1985; 29: 387-409.
validate the hypothesis of Cunningham et al.6 in
12. Takamura K, Okishima T, Ohdo S, Hayakawa K.
terms of a possibly new genetic syndrome.
Association of cephalic neural crest cells with cardiovascular development, particularly that of the
To the best of our knowledge, this is the first
semilunar valves. Anat Embryol 1990; 182: 263-272.
case in the literature of ARS with coexisting
13. Semina EV, Reiter R, Leysens NJ, et al. Cloning and
TA. In light of this association, we suggest that
characterization of a novel bicoid-related homeobox
the diagnosis of ARS should be followed by
transcription factor gene, RIEG, involved in Rieger
systemic evaluation for congenital heart diseases.
syndrome. Nat Genet 1996; 14: 392-399.
In patients with anterior segment dysgenesis and
14. Nishimura DY, Swiderski RE, Alward WL, et al. The
TA, analysis of the genes that cause anterior
forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat
segment dysgenesis or other related genes should
be pursued to determine their possible role in the pathogenesis of this syndrome.
15. Phillips JC, del Bono EA, Haines JL, et al. A second
locus for Rieger syndrome maps to chromosome 13q14. Am J Hum Genet 1996; 59: 613–619. REFERENCES
1. Shields MB. Axenfeld-Rieger syndrome: a theory of
16. Velinov M, Gu H, Yeboa K, et al. Hypoplastic left
mechanism and distinctions from the iridocorneal
heart in a female infant with partial trisomy 4q due
endothelial syndrome. Trans Am Ophthalmol Soc
to de novo 4;21 translocation. Am J Med Genet 2002;
2. Pearce WG, Wyatt HT, Boyd TA, Ombres RS, Salter
17. Mirza G, Williams RR, Mohammed S, et al.
AB. Autosomal dominant iridogoniodysgenesis: genetic
Refined genotype-phenotype correlations in cases of
features. Can J Ophthalmol 1983; 18: 7-10.
chromosome 6p deletion syndromes. Eur J Hum Genet 2004; 12: 718-728.
3. Shields MB. Axenfeld-Rieger and iridocorneal
endothelial syndromes: two spectra of disease with
18. Pont SJ, Robbin JM, Bird TM, et al. Congenital
striking similarities and differences. J Glaucoma 2001;
malformations among liveborn infants with trisomies 18
and 13. Am J Med Genet A 2006; 140: 1749-1756.
Module 2 Handout Evidence-Based Treatments (EBTs) for Depression What are Evidence-Based Treatments (EBT)? • EBTs apply the best available evidence gained from scientific research to decision-making about treatment. • EBTs are shown in randomized clinical trials to be effective in reducing symptoms of a particular disorder and/or improving functioning Types of EBTs for Depres
INTERDISCIPLINARY RESEARCH PROJECT FUNDED BY THE SPECIALISED COMMITTEE INTERDISCIPLINARY RESEARCH OF THE SWISS NATIONAL SCIENCE FOUNDATION (SNSF) Children’s Rights Unit PO Box 4176 • 1950 Sion 4 • Switzerland Tel. +41 27 205 73 00 • Fax +41 27 205 73 01 MAIN FINDINGS • The international priorities of child rights advocacy have significantly evolved over the period under revi

 Axenfeld-Rieger Syndrome with Truncus Arteriosus 445
examination displayed a prominent, anteriorly
latanoprost 0.005% (Xalatan, Pharmacia &
displaced Schwalbe’s line in all quadrants of
Upjohn, Uppsala, Sweden) once daily at 10:00
both eyes. The iris had stromal hypoplasia
pm, and after one month of therapy, intraocular
bilaterally. There were corectopia in the right
pressure was 18 mmHg in the left eye.
Axenfeld-Rieger Syndrome with Truncus Arteriosus 445
examination displayed a prominent, anteriorly
latanoprost 0.005% (Xalatan, Pharmacia &
displaced Schwalbe’s line in all quadrants of
Upjohn, Uppsala, Sweden) once daily at 10:00
both eyes. The iris had stromal hypoplasia
pm, and after one month of therapy, intraocular
bilaterally. There were corectopia in the right
pressure was 18 mmHg in the left eye.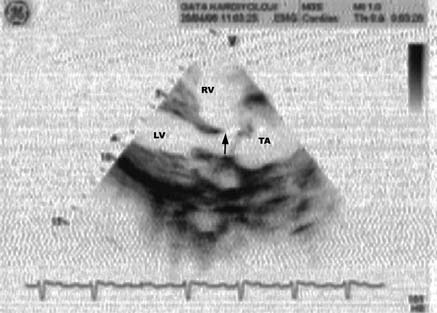
 The Turkish Journal of Pediatrics • October - December 2007
Owing to clubbing and history of shortness
ARS and TA, the range of reported cardiac
of breath, the patient was referred to a
malformations is enlarged and the importance
cardiologist, who found a soft systolic thrill
of cardiologic evaluation is emphasized in
along the left sternal border and loud apical
pansystolic murmur at the left lower sternal
border, radiating to the whole precordial
accompanying ARS have been described. In
area and especially to the right side. A two-
1994, Tsai et al.5 described a patient with aortic
dimensional echocardiographic examination
stenosis associated with ARS. Cunningham
revealed TA (Figs. 5 and 6). The patient was
et al.6 then described ARS coexisting with
admitted to another hospital for cardiac surgical
atrial septal defects and sensorineural hearing
loss, affecting multiple members of a single family. In 2000 Bekir et al.7 reported a 20-year-old girl with ARS associated with an atrial septal defect and interatrial aneurysm. Recently, Baruch and Erickson8 described two siblings presenting with ARS, hypertelorism, clinodactyly, and cardiac anomalies such as patent ductus arteriosus and atrial septal defect. Most recently, Grosso et al.9 reported a family with a clinical picture overlapping that described by Cunningham et al.6 and characterized by ARS in association with cardiac malformations and sensorineural deafness, without facial dysmorphisms, dental hypoplasia, or involuted periumbilical skin. However, in their patients,
Fig. 5. Echocardiographic characterization of truncus
The Turkish Journal of Pediatrics • October - December 2007
Owing to clubbing and history of shortness
ARS and TA, the range of reported cardiac
of breath, the patient was referred to a
malformations is enlarged and the importance
cardiologist, who found a soft systolic thrill
of cardiologic evaluation is emphasized in
along the left sternal border and loud apical
pansystolic murmur at the left lower sternal
border, radiating to the whole precordial
accompanying ARS have been described. In
area and especially to the right side. A two-
1994, Tsai et al.5 described a patient with aortic
dimensional echocardiographic examination
stenosis associated with ARS. Cunningham
revealed TA (Figs. 5 and 6). The patient was
et al.6 then described ARS coexisting with
admitted to another hospital for cardiac surgical
atrial septal defects and sensorineural hearing
loss, affecting multiple members of a single family. In 2000 Bekir et al.7 reported a 20-year-old girl with ARS associated with an atrial septal defect and interatrial aneurysm. Recently, Baruch and Erickson8 described two siblings presenting with ARS, hypertelorism, clinodactyly, and cardiac anomalies such as patent ductus arteriosus and atrial septal defect. Most recently, Grosso et al.9 reported a family with a clinical picture overlapping that described by Cunningham et al.6 and characterized by ARS in association with cardiac malformations and sensorineural deafness, without facial dysmorphisms, dental hypoplasia, or involuted periumbilical skin. However, in their patients,
Fig. 5. Echocardiographic characterization of truncus