Deracemisation of [alpha]-chiral primary amines by a one-pot, two-step cascade reaction catalysed by [omega]-transaminases
FULL PAPER DOI: 10.1002/ejoc.200801265 Deracemisation of α-Chiral Primary Amines by a One-Pot, Two-Step Cascade Reaction Catalysed by ω-Transaminases Dominik Koszelewski,[a] Dorina Clay,[a] David Rozzell,[b] and Wolfgang Kroutil*[a] Dedicated to Kalle Hult on the occasion of his 65th birthdayKeywords: Amines / Deracemisation / Asymmetric catalysis / Biotransformations / Enzymes
Racemic α-chiral primary amines were deracemised to op-
donor and a ω-transaminase displaying opposite stereopref-
tically pure amines in up to Ͼ99 % conversion and Ͼ99 % ee
erence than the ω-transaminase in the first step. In the sec-
within 48 h. The deracemisation was a result of a stereoinver-
ond step, lactate dehydrogenase was used to remove the side
sion of one amine enantiomer; the formal stereoinversion was
product pyruvate to shift the unfavourable reaction equilib-
achieved by a one-pot, two-step procedure: in the first step,
rium to the product side. Depending on the order of the en-
kinetic resolution of the chiral racemic amine was performed
antiocomplementary enzymes employed in the cascade, the
by employing a ω-transaminase to yield an intermediate
(R), as well as the (S), enantiomer was accessible.
ketone and the remaining optically pure amine; in the secondstep, the ketone intermediate was stereoselectively trans-
( Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim,
formed into the amine by employing alanine as the amine
Introduction
binding of the (R)-mexiletine to a cardiac sodium channeland the higher antiarrhythmic activity of this enantio-
Optically active amines are required for the preparation
mer.[12,13] Consequently, there is a need for efficient meth-
of a broad range of biologically active compounds showing
ods to obtain the desired (R) or (S) enantiomer in optically
various pharmacological properties.[1,2] Subsequently, chiral
pure form starting from easily accessible substrates. Resolu-
amines have been used as resolving agents, chiral auxiliaries
tion of some of these amines has been carried out by frac-
and building blocks in the synthesis of neurological, cardio-
tional crystallisation or distillation of the diastereomeric
vascular, immunological, antihypertensive, anti-infective
salts,[14,15] chromatographic separation of diastereomeric
and antiemetic drugs.[3] In most cases, the pharmacological
amides,[16] microbial hydrolysis of an N-acyl derivative[17–19]
activities of these amines are related to the configuration of
or by enantioselective acylation of racemic amines catalysed
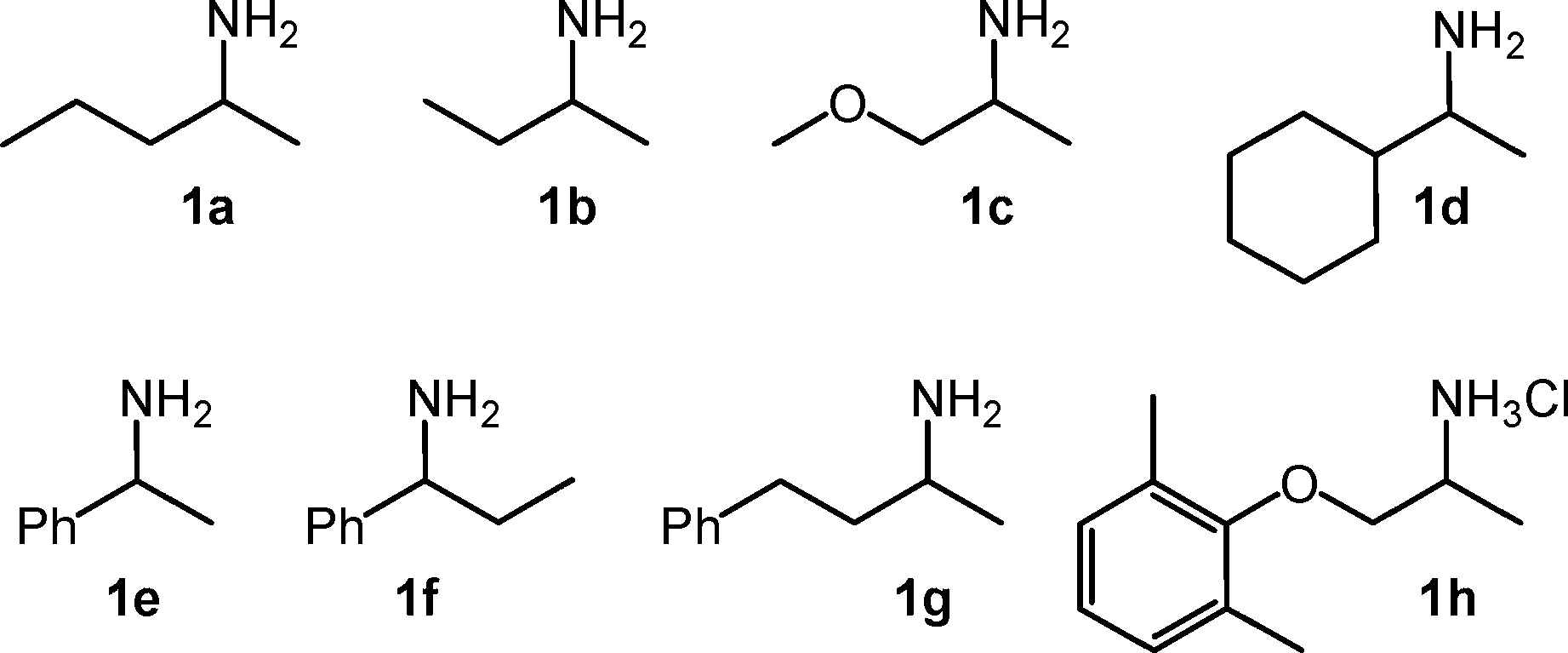
the stereogenic centre.[4,5] For example, (R)-4-phenylbutan-
by lipase B from Candida antarctica.[20–22] Furthermore, ω-
2-amine (1g) is a precursor of the antihypertensive dileva-
transaminases received recently increased attention for (i)
lol,[6] and (S)-sec-butylamine (1b), (S)-1-methoxy-2-propan-
the kinetic resolution of racemic amines and (ii) the asym-
amine (1c) and (S)-1-cyclohexylethylamine (1d) are precur-
metric amination of the corresponding ketones.[23–34]
sors of inhibitors of tumour necrosis factor-α(TNF-α).[7] Furthermore, 1-phenyl-1-propylamine (1f) is a precursor of corticotropin releasing factor type-1 receptor antagonist, a potent antidepressant agent.[8] Finally, mexiletine (1h) is an orally effective antiarrhythmic,[9] antimyotonic[10] and anal- gesic[11] agent in its racemic form and is available for clinical use as the racemate (Figure 1). Mexiletine undergoes stereo- selective disposition in vivo associated with the selective
[a] Research Centre Applied Biocatalysis c/o Department of Chem-
istry, Organic and Bioorganic Chemistry, University of GrazHeinrichstrasse 28, 8010 Graz, Austria
Figure 1. Racemic amines 1a–h chosen for the biocatalytic one-pot,
two-step deracemisation protocol employing two ω-transaminases.
Additionally, protocols like dynamic kinetic resolution of
Redwood City, California, USASupporting information for this article is available on the
amines[35–37] and cyclic deracemisation[38] for the deracemi-
WWW under http://www.eurjoc.org/ or from the author.
sation of racemic amines to yield optically pure products in
Eur. J. Org. Chem. 2009, 2289–2292
2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
D. Koszelewski, D. Clay, D. Rozzell, W. Kroutil
FULL PAPER
100 % yield and Ͼ99 % ee have been developed. Because in
Table 1. Kinetic resolution of amines 1 catalysed by commercially
various cases the racemic amine is more readily available
than the corresponding ketone, deracemisation will gain in-
creased attention; for example, the corresponding ketones
of 1d and 1h are not commercially available. Therefore, we
envisaged a two-step, one-pot process as outlined in
Scheme 1 that consists of (i) a kinetic resolution and (ii) a
stereoselective amination. The advantage of this concept is
to avoid the limitation of kinetic resolution (50 % conver-
sion) giving quantitative yield of optically pure amine start-
Results and Discussion
For the deracemisation of amines by a two-step process
as outlined in Scheme 1 a kinetic resolution step is required
first, followed by a reductive amination step.
[a] Reaction conditions: amine 1 (50 m), sodium pyruvate (50 m), ω-transaminase (6 mg), phosphate buffer (100 m, pH 7.0, 1 m PLP), shaking at 30 °C for 24 h. [b] Determined by GC. [c] Determined by GC analysis on a chiral phase. [d] A 25 m solu-
Scheme 1. One-pot, two-step synthesis procedure towards enantio-
tion of sodium pyruvate was employed. [e] 30 m.
enriched amines employing two ω-transaminases (ω-ATAs) withopposite stereopreference. Removal of pyruvate was performed byits reduction employing lactate dehydrogenase (LDH) to shift theequilibrium to the product side.
(Scheme 1). Shifting the ketone–amine equilibrium to the
Testing first the kinetic resolution for various amines,
amine side is a challenge with ω-transaminases, especially
three commercial ω-transaminases (ATA-113, ATA-114
when using an amino acid like alanine as an amino donor,
and ATA-117) were chosen as catalysts (Table 1). The ω-
as in this case the equilibrium is on the side of the sub-
transaminases catalysed this reaction efficiently, giving the
strates (ketone, alanine) and not on the side of the products
amines with up to Ͼ99 % ee (Table 1).
(amine, pyruvate).[23] To shift the equilibrium[29,31] the pyr-
For a number of substrate/enzyme combinations, the for-
uvate formed was removed by reduction by using lactate
mal ideal 50 % conversion barrier was surpassed, indicating
dehydrogenase (LDH) in a coupled reaction system. The
that the ω-transaminases possess for these amines a nonper-
stereoselectivity for the reductive amination of the commer-
fect enantioselectivity. For instance, in the case for which a
cial ketones was already previously reported.[24]
limiting amount of pyruvate was used, the enantiomeric ex-
The reaction sequence was performed in a way that after
cess at 50 % conversion was not perfect (Table 1, Entries 2
the kinetic resolution the second ω-transaminase with op-
and 4). However, because the ketone will be transformed
posite stereopreference was added together with the corre-
back to the remaining amine enantiomer in the second step
sponding alanine enantiomer. Because the two ω-transami-
of the one-pot transformation, the goal of the first step is
nases should also have different stereopreference for the ala-
complete removal of the “wrong” enantiomer. The most im-
nine enantiomers, we speculated that this approach should
portant point, however, was that transaminases ATA-113
work. However, as can be seen from the results (Table 2),
and ATA-114 showed (S) preference, whereas ATA-117 dis-
the optical purity of the final product was moderate, al-
played (R) preference; thus, enantiocomplementary en-
though exclusively amine could be detected in almost all
zymes were available. This is actually the basis for the com-
cases. The reason for this was that the ω-transaminases also
accept, to a certain extent, the opposite alanine enantiomer,
Having identified the suitable conditions for the kinetic
meaning that the ω-transaminase of the first step also cata-
resolution of the desired amines we turned our attention to
lysed the amination reaction although at a reduced rate.
couple the first step (i.e., kinetic resolution) with the stereo-
Overall, this led to a diminished ee value. The same was
selective amination to achieve a deracemisation reaction se-
observed when DMSO was added in the second step, al-
2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Eur. J. Org. Chem. 2009, 2289–2292
Deracemisation of α-Chiral Primary Amines
though DMSO partially inhibited ω-transaminase ATA-
Depending on the order of addition of the ω-transami-
113, whereas the activity of ATA-117 stayed untouched
nases, either the (R) or the (S) enantiomer was accessible.
(Table 2, Entries 1 and 3 vs. 2 and 4).
Thus, just by switching the order, the other enantiomer wasobtained.
Table 2. Two-step procedure catalysed by various commercially
Employing the optimised conditions, a preparative trans-
formation of 50 mg of rac-1d at 20 m substrate concentra-
tion yielded (S)-1d at Ͼ99 % conversion after 48 h with
Ͼ99%ee and 92% isolated yield. Transformation of 50 mg
of racemic 1-(2,6-dimethylphenoxy)-2-propanamine (1h) led
to optically pure amine (S)-1h with complete conversion,
Ͼ99%ee and 95% isolated yield. Conclusions
[a] Order of addition of ω-transaminase; in the case of ATA-117,
-Ala was used in the amination reaction and -Ala was used with
In summary, a one-pot, two-step deracemisation cascade
ATA-113 and ATA-114. [b] Determined by GC. [c] Determined byGC analysis on a chiral phase.
leading to optically pure pharmacologically relevant aminesthrough kinetic resolution and subsequent stereoselective
To solve this problem, heat treatment was introduced be-
amination catalysed by two enantiocomplementary ω-trans-
tween the two steps. Thus, after the kinetic resolution, the
aminases was described. The resulting amines were ob-
sample was kept at 75 °C for 30 min before the enzyme re-
tained with up to quantitative conversion and excellent
quired for the second step was added. Although destruction
enantioselectivities (up to Ͼ99 % ee).
of the enzyme may lead to an expensive process, this trickenabled efficient one-pot, two-step deracemisation leadingto optically enriched and even optically pure (R)- or (S)-
Experimental Section
amines with very high conversion (Table 3). From all eight
Amines 1a–h and ketones, as well as solvents (DMSO), were pur-
substrates 1a–h only the (S) enantiomer of 1g was obtained
chased from Sigma–Aldrich (Vienna, Austria) or BASF (Ludwigsh-
with a low ee value. For all other cases, the ee value was at
afen, Germany) and used as received unless otherwise stated. All
least 96 % [(R)-1c], otherwise the value was Ͼ99 % for both
chemicals used were of analytical grade. ω-Transaminases ATA-
0.46 U mg–1; transaminase ATA-117, 102907WW, 1.9 U mg–1;transaminase ATA-114, 1091108MW, 2.7 U mg–1) and lactate dehy-
Table 3. Two-step procedure with the addition of heat to 75 °C after
drogenase mix (LDH, PRM-102, 101807KVP, mixture of lactate
the first step catalysed by various commercially available ω-trans-
dehydrogenase, glucose dehydrogenase, glucose, NAD+) were ob-
tained from Codexis Inc. One unit of ω-transaminase was defined
as the amount of enzyme that catalyses the formation of 1 µmolacetophenone from α-methylbenzylamine at pH 9.0 at 22 °C. Kinetic Resolution: All biocatalytic reactions were performed at
30 °C for 24 h in sodium phosphate buffer (100 m, pH 7) contain-
ing pyridoxal-5Ј-phosphate monohydrate (1 m) in a 2-mL eppen-
dorf tube. The reaction buffer (1 mL) was mixed with ω-transami-
nase (6 mg) and sodium pyruvate (50 m). The reaction mixture
contained 50 m of corresponding amine 1. The conversion to
ketone 2 was measured by GC chromatography. The reaction was
stopped by adding NaOH (200 µL, 10 ), followed by extraction
with ethyl acetate (600 µL, 2ϫ). Organic phase was dried with an-
Analysis of Optical Purity of Products: The enantiomeric excess of
amines 2a–g was analysed by gas chromatography on a chiral phase
after derivatisation to the acetoamides, which was performed by
adding a DMAP and a 20-fold excess of acetic acid anhydride.
Amine 2h was analysed after derivatisation to trifluoroacetamide,
which was performed by adding a 20-fold excess of trifluoroacetic
acid anhydride. After washing with water and drying with anhy-
2SO4 the ee value of the derivatised compound was mea-
[a] Order of addition of ω-transaminase; in the case of ATA-117,
Representative Example for Amination with Heating at 75 °C: After
-Ala was used in the amination reaction and -Ala was used with
the kinetic resolution step (24 h), the mixture was kept at 75 °C for
ATA-113 and ATA-114. [b] Determined by GC. [c] Determined by
30 min, cooled to room temperature and LDH-mix (40 mg, 1 m
NAD+, glucose, lactate dehydrogenase, glucose dehydrogenase) was
Eur. J. Org. Chem. 2009, 2289–2292
2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
D. Koszelewski, D. Clay, D. Rozzell, W. Kroutil
FULL PAPER
added (to remove pyruvate from the first step), and the mixture
[15] M. Acs, T. Szili, E. Fogassy, Tetrahedron Lett. 1991, 32, 7325–
was shaken for 1 h at 30 °C. - or -Alanine (5 equiv., 250 m)
and the second ω-transaminase (6 mg, Table 3) were added, and
[16] R. Aav, O. Parve, T. Pehk, A. Claesson, I. Martin, Tetrahedron:Asymmetry 1999, 10, 3033–3038.
the mixture was shaken at 30 °C. After 24 h, 200 µL 10 NaOH
[17] S. Buchholz, H. Gröger in Biocatalysis in the Pharmaceutical
and 600 µL EtOAc were added. The organic phase was dried
and Biotechnology Industries (Ed.: R. N. Patel), CRC Press,
(Na2SO4) and analysed by GC chromatography.[24,25]
Boca Raton, 2007, pp. 829–847. Supporting Information (see footnote on the first page of this arti-
[18] M.-J. Kim, Y. Ahn, J. Park in Biocatalysis in the Pharmaceuti-cal and Biotechnology Industries (Ed.: R. N. Patel), CRC Press,
cle): Preparative transformations, determination of absolute config-
Boca Raton, 2007, pp. 249–272.
uration and (chiral) analytics are reported.
[19] J. Ogawa, S. Shimizu, H. Yamada, Bioorg. Med. Chem. 1994,
[20] B. Martin-Matute, J.-E. Bäckvall, Curr. Opin. Chem. Biol. 2007, Acknowledgments
[21] J. González-Sabín, V. Gotor, F. Rebolledo, Tetrahedron: Asym-
Financial support by the Österreichische Forschungsförderungsge-
metry 2002, 13, 1315–1320.
sellschaft (FFG) and the Province of Styria is gratefully acknowl-
[22] V. Gotor-Fernández, E. Busto, V. Gotor, Adv. Synth. Catal.
edged. Codexis is thanked for providing various enzymes. 2006, 348, 797–812.
[23] M. Höhne, S. Kühl, K. Robins, U. T. Bornscheuer, ChemBio-Chem 2008, 9, 363–365.
[1] H. Y. Aboul-Enein, I. W. Wainer, The Impact of Stereochemis-
[24] D. Koszelewski, I. Lavandera, D. Clay, D. Rozzell, W. Kroutil,
try on Drug Development and Use, Wiley, New York, 1997. Adv. Synth. Catal. 2008, 350, 2761–2766.
[2] E. J. Ariëns, W. Soudijn, P. B. M. W. M. Timmermans, Stereo-
[25] D. Koszelewski, I. Lavandera, D. Clay, D. Rozzell, W. Kroutil,
chemistry and Biological Activity of Drugs, Blackwell, Oxford,
Angew. Chem. Int. Ed. 2008, 47, 9337–9340; Angew. Chem. 2008, 120, 9477–9480.
[3] L. Sutin, S. Andersson, L. Bergquist, V. M. Castro, E. Dan-
[26] S.-S. Yi, C.-w. Lee, J. Kim, D. Kyung, B.-G. Kim, Y.-S. Lee,
ielsson, S. James, M. Henriksson, L. Johansson, C. Kaiser, K. Process Biochem. 2007, 42, 895–898.
Flyrén, M. Williams, Bioorg. Med. Chem. Lett. 2007, 17, 4837–
[27] U. Kaulmann, K. Smithies, M. E. B. Smith, H. C. Hailes, J. M.
Ward, Enzyme Microb. Technol. 2007, 41, 628–637.
[4] S. H. Snyder, K. M. Tayler, Science 1970, 168, 1487–1489.
[28] B.-Y. Hwang, B.-K. Cho, H. Yun, K. Koteshwar, B.-G. Kim,
[5] G. T. Hajos, S. Garattin, J. Pharm. Pharmacol. 1973, 25, 418– J. Mol. Catal. B: Enzym. 2005, 37, 47–55.
[29] H. Yun, B.-Y. Hwang, J.-H. Lee, B.-G. Kim, Appl. Environ.
[6] J. E. Clifton, I. Collins, P. Hallett, D. Hartley, L. H. C. Lunts,
Microbiol. 2005, 71, 4220–4224.
P. D. Wicks, J. Med. Chem. 1982, 25, 670–679.
[30] H. Yun, S. Lim, B.-K. Cho, B.-G. Kim, Appl. Environ. Micro-
[7] M. P. Clark, S. K. Laughlin, M. J. Laufersweiler, R. G. Book-
biol. 2004, 70, 2529–2534.
land, T. A. Brugel, A. Golebiowski, M. P. Sabat, J. A. Townes,
[31] J.-S. Shin, B.-G. Kim, Biotechnol. Bioeng. 2002, 77, 832–837.
J. C. VanRens, J. F. Djung, M. G. Natchus, B. De, L. C. Hsieh,
[32] J. Ager, T. Li, D. P. Pantaleone, R. F. Senkpeil, P. P. Taylor,
S. C. Xu, R. L. Walter, M. J. Mekel, S. A. Heitmeyer, K. K.
I. G. Fotheringham, J. Mol. Catal. B: Enzym. 2001, 11, 199–
Brown, K. Juergens, Y. O. Taiwo, M. J. Janusz, J. Med. Chem.2004, 47, 2724–2727.
[33] J. D. Stewart, Curr. Opin. Chem. Biol. 2001, 5, 120–129.
[8] J. W. Corbett, M. R. Rauckhorst, F. Qian, R. L. Hoffman,
[34] M. D. Truppo, J. D. Rozzell, J. C. Moore, N. J. Turner, Org. Bi-
C. S. Knauer, L. W. Fitzgerald, Bioorg. Med. Chem. Lett. 2007, omol. Chem. 2009, 7, 395–398.
[35] C. E. Hoben, L. Kanupp, J.-E. Bäckvall, Tetrahedron Lett.
[9] P. E. Fenster, K. A. Comess, Pharmacotherapy 1986, 6, 1–9. 2008, 49, 977–979.
[10] R. Rudel, F. Lehmann-Horn, Physiol. Rev. 1985, 65, 310–356.
[36] A. N. Parvulescu, P. A. Jacobs, D. E. De Vos, Adv. Synth. Ca-
[11] E. Kalso, M. R. Tramer, H. J. McQuay, R. A. Moore, Eur. J.tal. 2008, 350, 113–121. Pain 1998, 2, 3–14.
[37] A. N. Parvulescu, P. A. Jacobs, D. E. De Vos, Chem. Eur. J.
[12] C. Franchini, C. Cellucci, F. Corbo, G. Lentini, A. Scilimati,
2007, 13, 2034–2043.
V. Tortorella, F. Stasi, Chirality 1994, 6, 590–595.
[38] N. J. Turner, R. Carr in Biocatalysis in the Pharmaceutical and
[13] A. Carocci, C. Franchini, G. Lentini, F. Loiodice, V. Tortorella,
Biotechnology Industries (Ed.: R. N. Patel), CRC Press, Boca
Chirality 2000, 12, 103–106.
Raton, 2007, pp. 743–755.
[14] C. Franchini, C. Cellucci, F. Corbo, G. Lentini, A. Scilimati,
V. Tortorella, F. Stasi, Chirality 1994, 6, 590–595.
2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Eur. J. Org. Chem. 2009, 2289–2292
Equity Portfolio as at 30 June 2006 Security Name Value in New Zealand Dollars Total Argentina 1,025,970 AUSTRALIA AND NEW ZEALAND BANKING GROUP LIMITED New Zealand Superannuation Fund 6 September 2006 - Page 1 of 52 Equity Portfolio as at 30 June 2006 Security Name Value in New Zealand Dollars MACQUARIE GOODMAN GROUP STAPLED DEFERRED SET New Zealand Superannuation Fu
«Original Articles » Comparison of therapeutic abortion efficacy by suction curettage and misoprostol vaginally in the first trimester of pregnancy Mahvash Zargar 1, Roshan Nikbakht 1, Vida Naji givi 1, Masoud Hemadi1* Abstract Infertility and Perinatology Research Background: This study compared the health outcomes of abortion Center, Ahvaz Jundishapur University by vagi


 FULL PAPER
FULL PAPER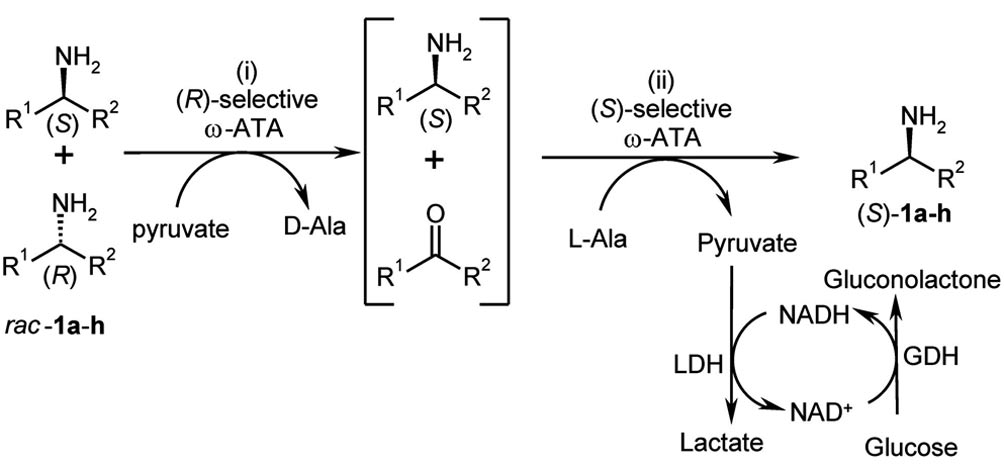 D. Koszelewski, D. Clay, D. Rozzell, W. Kroutil
FULL PAPER
D. Koszelewski, D. Clay, D. Rozzell, W. Kroutil
FULL PAPER







 Deracemisation of α-Chiral Primary Amines
though DMSO partially inhibited ω-transaminase ATA-
Depending on the order of addition of the ω-transami-
113, whereas the activity of ATA-117 stayed untouched
nases, either the (R) or the (S) enantiomer was accessible.
Deracemisation of α-Chiral Primary Amines
though DMSO partially inhibited ω-transaminase ATA-
Depending on the order of addition of the ω-transami-
113, whereas the activity of ATA-117 stayed untouched
nases, either the (R) or the (S) enantiomer was accessible.