General introduction Homogeneous catalysis with metal phosphine complexes
Tertiary organic phosphines (PR3; R= alkyl, aryl) were discoveredaround the middle of the 19th century. Their ability to combinewith heavy metal salts was noted almost immediately, but theapplication of the metal complexes in homogeneous catalysis is adevelopment which only started to flourish after the 1950s.1The first event of direct relevance to the application of phosphinecomplexes to catalysis occurred immediately after the secondWorld War. Reppe and coworkers showed that varioustriphenylphosphine complexes of nickel (especially(PPh3)2Ni(CO)2) were more effective than other nickel complexesfor the polymerization of olefinic and acetylenic substances. Othernickel complexes, especially (PPh3)2NiBr2, catalyzed the formationof acrylic acid esters from alcohols, acetylene and carbonmonoxide.2,3 The mechanisms of these catalytic reactions wereobscure, but the demonstration that phosphine complexes hadpotentially useful catalytic properties attracted the attention of thepetrochemical industry world-wide. Another major discovery was that of RhCl(PPh3)3 (Wilkinson’scatalyst) in 1965, a phosphorus modified homogeneous catalyst forthe hydrogenation of alkenes at room temperature andatmospheric pressure of H2.4 Its remarkable activity and selectivitycompared to heterogeneous systems resulted in investigation ofrelated complexes, such as RuHCl(PPh3)3, today one of the mostactive homogeneous catalysts for the hydrogenation of 1-alkenes.5Furthermore, investigations aiming at optimization of this reactionled to the understanding that the properties of the phosphine used
is important for the selectivity. These experiments led to a generalappreciation of the fact that changing substituents of phosphorusligands, and thus changing the electronic and/or steric influence ofthe ligands, can cause marked changes in the behavior of theirtransition metal complexes. Tolman has defined an electronicparameter χ for phosphorus ligands based upon the difference inthe IR frequencies of Ni(CO)3L and the reference compoundNi(CO)3(PtBu3). The variability of the phosphorus ligands is nicelyreflected in the IR frequencies, which can be measured withsufficient accuracy to give different values for the relevantsubstituents. Alkyl and aryl substituents give values ranging from 0to 20 cm-1, phosphites (alkoxy substituents) from 20 to 40 cm-1,and halogen substituents give values up to 59 cm-1. A ligand with ahigh χ-value is electron-withdrawing and easily accepts π -backbonding from the metal; a ligand such as PF3, which is at thetop of the range, is a strong π-acceptor like CO. The most widely accepted steric parameter is θ, the cone angle, alsointroduced by Tolman.6 The cone angle θ is defined as the apexangle of a cylindrical cone, centered at 2.28 Å from the center ofthe P-atom, which just touches the Van der Waals radii of theoutermost atoms of the model (see Figure 1). If there are internaldegrees of freedom, the substituents are folded in such a way thata minimally sized cone is formed. Figure 1. Illustration of the cone angle
Crystal structure determinations have shown that in practice theangles found in the structures are smaller than is suggested by theθ value. For example, two cis triphenylphosphine ligands may havea P–M–P angle as small as 95°, whereas the θ values would predict145°. In reality intermeshing of the substituents on the phosphorusleads to smaller cone angles. Originally the cone angles ofphosphine ligands were determined by measurements on the CPKmodel, but recently some more advanced methods involvingmolecular mechanics have been applied.7-9The χ and θ parameters have to be used with caution, since fullseparation of the steric and electronic parameters is not possible. Steric interactions might bend the alkyl groups away from the idealangles, destabilize their bonding σ orbitals and lower theantibonding π * orbitals, thus enhancing π-backdonation andconsequently the electronic influence.
The greater stability of complexes of bidentate ligands comparedwith the corresponding complexes containing similar monodentateligands is well known and commonly referred to as the “chelateeffect”. The nature of this effect has been explained in terms of theentropy change involved in the chelation process.10 After thediscovery and application of diamines in 1889 (Jørgenson), the α-diimines 2,2’-bipyridine and o-phenanthroline in 1898 (Blau), thediarsine o-phenylenebisdimethylarsine in 1939 (Chatt and Mann;Nyholm), the first diphosphine 1,2-bis(diphenylphosphino)ethane(dppe) was synthesized by Wymore and Bailar in 1956.11The stability of cis chelate complexes varies with the chelate ringsize, and reaches a maximum for five membered rings, such asthose formed by bidentate ligands with ethylene or ortho-phenylene bridges between the two donor atoms. Therefore thevast majority of chelate complexes have been synthesized frombidentate ligands possessing relatively short, bridging, backbones. Besides stabilizing the metal complex, the application of adiphosphine can have a marked influence on the selectivity of acatalyst as well. For example the carbonylation of ethene in
methanol, catalyzed by a phosphine/Pd(II) catalyst, leads to theselective formation (> 98%) of methylpropionate when PPh3 is usedas ligand. When 1,3-bis(diphenylphosphino)-propane (dppp) isemployed, a perfectly alternating copolymer of ethene and carbonmonoxide is formed with > 99% selectivity.12
Figure 2. Carbonylation of ethene
Even though the short bridges found in most diphosphine ligandsenhances the stability of a large number of complexes, they alsolimit their application. Many catalytic processes involve one ormore intermediates in their catalytic cycles with tetrahedral (∠P–M–P= ca. 109°) and/or trigonal bipyramidal geometries (∠P–M–P=ca. 120°). The short backbones in the common diphosphines arenot suitable to support the P–M–P angles in these geometries; thesmall chelate ring forces the metal into a conformation with P–M–Pangles of 90° or smaller angles. A means to predict chelational preferences of bidentate ligands wasrecently developed by Casey and Whiteker.13 They introduced theconcepts of the natural bite angle and the flexibility range fordiphosphine ligands, which can be calculated by molecularmechanics. The natural bite angle (βn) is defined as the preferredchelation angle determined only by ligand backbone constraintsand not by metal valence angles. The flexibility range is defined asthe accessible range of bite angles within less than 3 kcal.mol-1excess strain energy from the calculated natural bite angle. Comparison of different diphosphines as ligands in varioushomogeneously catalyzed reactions reveals that the application ofa diphosphine with a large natural bite angle can have dramatic
effects on the activity and selectivity induced by a catalyst. Forinstance Hayashi et al. showed that [dppf]PdCl2, a diphosphinewith a large P–Pd–P bite angle of 99.07°, induces very high activityand selectivity in the palladium-catalyzed cross-coupling of 2-butylmagnesiumchloride with bromobenzene, whereas application of[dppe]PdCl2 (with a P–Pd–P angle of 85.8°14) leads to no conversionat all.15,16
Casey et al. showed that in the rhodium-catalyzedhydroformylation the natural bite angle of the diphosphine ligandused has a dramatic influence on the regioselectivity of thereaction. BISBI (2,2’-bis((diphenylphosphino)methyl)-1,1’-biphenyl), a ligand that was shown to coordinate in a bisequatorialfashion (i.e. ∠P–Rh–P= ca. 120°) induces a ratio of linear tobranched aldehyde of up to 66 : 1, whereas dppe, whichcoordinates in a equatorial-axial fashion (i.e. ∠P–Rh–P= ca. 90°)leads to a linear to branched ratio of 2.1 : 1.8
Figure 2. Rhodium catalyzed hydroformylation Diphosphine ligands with large bite angles
The first reported example of a diphosphine with a large bite anglewas that in the trans complex [Ni(cHex)2P(CH2)5P(cHex)2Cl2],synthesized by Issleib and Hohlfield in 1961,17 during their
investigations of the formation of large chelate rings. They foundcomplexes containing a 5-, 6-, or 7-membered chelate ring to formexclusively cis-complexes, while the strainless 8-membered chelatering in [Ni(cHex)2P(CH2)5P(cHex)2Cl2] was found to be a trans-complex. The relation of chelate ring size and complex formation wasfurther investigated by Shaw, who synthesized a whole series oflong-chain diphosphine ligands of the type [tBu2P(CH2)nPtBu2],with n= 5 to 12. The formation of mononuclear or oligonuclearcomplexes was found to depend strongly on the size of thechelating ring. Stable mononuclear complexes where found with n=10 (Ir(tBu2P(CH2)10PtBu2)(CO)Cl, a 13-atom ring) and n=12(Ir(tBu2P(CH2)12PtBu2)(CO)Cl),18,19 although a dinuclear complexof the former compound was also observed. Similar diphosphineligands with longer or shorter chain lengths were found to giveoligonuclear structures20 or metallated products.21 The transchelating behaviour of the long chain bisphosphines is induced bythe bulky t-butyl groups; the formation of cis complexes ishindered by the steric hindrance of the t-butyl groups. Theformation of trans complexes of this type of long-chaindiphosphines proved to be very dependent on the complexprecursor and the reaction conditions used.22 Unless a very dilutedsolution is used, most of these ligands will coordinate preferably asa bridging ligand, rather than as a chelating ligand. These ligandshave found very little application other than in coordinationchemistry, since even though this type of diphosphines is able tosupport large bite angles, they are not able to enforce large biteangles.
The first ligand designed to enforce trans chelation was 2,11-bis-(diphenylphosphinomethyl)benzo(c)phenanthrene, TRANSPHOS,by Venanzi and coworkers.23 This ligand contains a rigidpolyaromatic backbone, forcing two methylene diphenylphosphinemoieties in positions allowing the formation of trans chelates (seebelow). The ligand gave trans chelated complexes with almost all
middle and late transition metals (Ni, Pd, Pt,24 Cu, Ag, Au,25,26 Rh,Ir,27-29 Fe, Ru30) with varying P–M–P angles. In the Cu (131.9˚) andAg (139˚ to 167.6˚31) complexes the deviation from the ideal 180˚was found to be very large, and with Pt(TRANSPHOS) even a few ciscomplexes were obtained32 (P–M–P angle of 104.8˚), indicating anunexpected flexibility of this ligand. TRANSPHOS Marty's POP ligand
An analogue of TRANSPHOS was synthesized by Marty and coworkers. Their POP ligand (3,3’-oxybis((diphenylphosphino)- methyl)benzene) contains a less rigid backbone than the benzo(c)phenanthrene unit used by Venanzi. The coordination chemistry of the ligand was very similar.33 Gillie and Stille showed that direct 1,1 reductive elimination from a cis (dppe)PdMe2 complex is a feasible reaction step, but a trans- dimethylpalladium complex containing TRANSPHOS resisted reductive elimination, even when heated to 100 °C. However, upon addition of CD3I rapid reductive elimination took place, and exclusively 1,1,1-trideutero-ethane was formed. They concluded that reductive elimination from this trans complex proceeds most likely from the five coordinate species [(TRANSPHOS)Pd(CH3)2(CD3)]I, which is formed after oxidative addition of CD3I to the palladium center.34 A ligand having a large bite angle, but not showing trans chelating behavior, 1,2-bis(diphenylphosphino)methyl-cyclopropane (TBDCP ), was developed by Rhône Poulenc.35 This ligand was used successfully in asymmetric hydrogenation. An X-ray crystal
structure of Fe(CO)3(TBDCP ) reported later by Casey and Whiteker showed a P–Fe–P angle of 123.9(1)˚.36 Rhodium catalyzed hydroformylation experiments with TBDCP were unsuccessful, due to the formation of a dimer upon warming to 25 ˚C (see below).37
Application of DIOP, a ligand designed for asymmetrichydrogenation,38 leads to relatively high regioselectivity in therhodium-catalyzed hydroformylation of 1-hexene (a linear tobranched aldehyde ratio of 8 : 1). For this diphosphine abisequatorial coordination (∠P–Rh–P= ca. 120°) is expected.39 P–M–P angles observed in X-ray crystal structures of transition metalcomplexes of DIOP range from 90.29(6)° in (DIOP)2Ru(H)(Cl)40 to106.4° in (DIOP)Pd(C2H4).41
For the ligand endo,endo-2,5-bis((diphenylphosphino)methyl)-bicyclo[2.2.1]heptane bisequatorial coordination was expected aswell,42 but application in the rhodium catalyzed hydroformylationdid not result in high selectivity. The observed linear to branchedratio was 2.6, like that found for monodentate PPh3, indicating thatthe ligand most probably does not form chelates.37 Application ofthis ligand in the rhodium catalyzed hydroformylation ofvinylarenes gave high regioselectivity under mild conditions, with
high branched to linear ratios (97:3 for styrene; 98:2 for 2-methoxy-6-vinyl-naphtalene, a precursor for naproxen).43
BISBI, as already mentioned, supports large bite angles, but israther flexible. X-ray structural analyses of transition metalcomplexes of BISBI show a wide variety of P-M-P chelation angles:∠P-Mo-P= 103.5° in (BISBI)Mo(CO)4,44 ∠P-Ir-P= 117.9° in(BISBI)Ir(H)(CO)2, ∠P-Rh-P= 124.8° in (BISBI)Rh(H)(CO)(PPh3),39and ∠P-Fe-P= 152.0° in (BISBI)Fe(CO)3.36
A new group of ligands designed to span trans positions are thechiral (R,R)-(S,S)-2,2”-bis[1-(dialkylphosphino)ethyl]-1,1”-biferrocene (TRAP) ligands,45 which have been used effectively inthe rhodium-catalyzed asymmetric Michael reaction46,47 and therhodium-catalyzed hydrosilylation.48
X-ray crystal structures reveal large P-M-P chelation angles:∠P-Pd-P= 163.6° in (Ph-TRAP)PdBr2, and ∠P-Rh-P= 161.06° in(FurTRAP)Rh(CO)Cl.49TRAP is not a very rigid ligand; (small) quantities of cis-complexesare observed in the 31P NMR of crude reaction mixtures of theplatinum complexes.45 This flexibility is probably due to freerotation around the link between the aromatic rings in thebackbone, as was also found with BISBI.39
Very recently, Trost has introduced several new diphosphineligands which are able to support large bite angles for use in theasymmetric allylic alkylation.50 An X-ray crystal structure of a
Pd[allyl] complex of the most selective of these ligands (with ananthracene-based backbone) shows a P–Pd–P bite angle of 110.5°. The high asymmetric induction was ascribed to this large biteangle.
Diphosphines with large bite angles can be interesting and usefulin many areas of homogeneous catalysis and coordinationchemistry. The results obtained with dppf, DIOP and BISBI haveshown that P–M–P bite angles in the range of 100 to 120° are veryattractive for various homogeneously catalyzed processes. Scope and contents of this thesis
This thesis deals with the development and application of newdiphosphine ligands, designed to induce large P–M–P angles intransition metal complexes. Aided by computational chemistry, ahomologous range of diphosphines based on rigid heterocyclicaromatic backbones of the xanthene-type with natural bite anglesof ca. 100° to ca. 134° have been developed.51
generic structure Sixantphos Thixantphos Xantphos
Substitution of the methylene bridge in the heterocyclic ring byother bridges allows a subtle alteration of the bite angle inducedby these ligands. The development and synthesis of these ligands isdescribed in Chapter 1.
Chapter 2 describes the application of these ligands in thecoordination chemistry of zerovalent palladium-TCNE complexes(TCNE= tetracyanoethylene). The application of this homologousrange of diphosphines is used to investigate the preferred biteangle in this type of complexes.
In Chapter 3 the application of the Xantphos-type of ligands in therhodium-catalyzed hydroformylation, an importanthomogeneously catalyzed process used in industry, is described. Comparison of the selectivities induced by known diphosphineshas lead to the assumption that the bite angle of diphosphines canbe of great importance for the selectivity towards the (industriallymore desirable) linear product. A large bite angle generally leadsto higher selectivities; the large bite angle induced by Xantphosleads to the highest selectivity reported for diphosphines so far. Owing to the rigidity of the backbones of the Xantphos-typeligands the selectivity is retained at higher temperatures, whichopens the possibility to obtain high selectivities at higher reactionrates.
The palladium-catalyzed carbon-carbon bond formation has foundextensive synthetic application. Even though the influence of thestabilizing (diphosphine) ligand on the activity and selectivity ofthe catalyst is known to be large, an explanation is still lacking. Comparison of the activities and selectivities induced byapplication of diphosphines inducing increasing bite angles cangive important information about the factors influencing theselectivity in these processes. In Chapter 4, investigations of thepalladium-catalyzed cross-coupling and allylic alkylation reactionsare described. The Xantphos-type ligands induce bite angles thathave not been explored before and their application in theseprocesses gives important information about the mechanism andfactors influencing selectivities.
The nickel-catalyzed hydrocyanation is an industrially importantroute to nitriles, which are used for instance for the production ofNylon-type polymers. The industrial importance of this reactionhas triggered a lot of research to design effective ligands that areable to stabilize the nickel catalyst under the corrosive reactionconditions involving hydrogen cyanide. Even though theapplication of phosphines and diphosphines has been investigatedby different research groups, the development of effectivephosphine-based catalysts has been unsuccessful so far. The mainreason is the instability of the nickel diphosphine intermediates inthis reaction. This leads to precipitation of insoluble nickeldicyanides, and consequently irreversible loss of catalyst. InChapter 5, the application of Xantphos-type ligands in the nickel-catalyzed hydrocyanation of styrene is described. Due to theirlarge bite angles these diphosphines are able to stabilizetetrahedral geometries and therefore these ligands do givesuccessful catalysis.
One of the most intriguing areas in modern coordination chemistryis the coordination chemistry of dihydrogen. The balance between
classical hydride complexes and non-classical dihydrogencomplexes is very subtle, and recent theoretical studies and studiesof the currently available (diphosphine)2RuH3+ complexes indicatethat especially at larger bite angles the steric effects of the ligandsbecome important. Calculations of possible geometries showed thatincreasing bite angles induce structural changes in(diphosphine)2Ru(“H3”)+ from trans hydride dihydrogen viadistorted cis hydride dihydrogen to trihydride complexes. Theapplication of diphosphines with larger bite angles than thosecurrently available has proven to be interesting for theinvestigation of the factors influencing the formation of hydridedihydrogen versus classical trihydride complexes. In Chapter 6 theapplication of the Xantphos-type diphosphines in the coordinationchemistry of dihydrogen is described. References
(1) Chatt, J. In Homogeneous Catalysis with Metal Phosphine Complexes;
Spignolet, L. H., Ed.; Plenum Press: New York, 1983; pp 1-11.
(2) Reppe, W.; Schweckendiek, W. J. Annalen 1948 , 560, 104-116. (3) Reppe, W.; Schweckendiek, W. J. German Patent 871, 494; Chem. Abstr. 1958 , 52, 10175.
(4) Young, J. F.; Osborn, J. A.; Jardine, F. H.; Wilkinson, G. J. Chem. Soc.,Chem. Commun. 1965 , 131-132.
(5) Evans, D.; Osborn, J. A.; Jardine, F. H.; Wilkinson, G. Nature (London)1965 , 208, 1203-1204.
(6) Tolman, C. A. Chem. Rev. 1977 , 77, 313-348. (7) DeSanto, J. T.; Mosbo, J. A.; Storhoff, B. N.; Bock, P. L.; Boss, R. E. Inorg. Chem. 1980 , 19, 3086.
(8) Chin, M.; Durst, G. L.; Head, S. R.; Bock, P. L.; Mosbo, J. A. J. Organomet.Chem. 1994 , 470, 73-85.
(9) Dias, P. B.; Minas de Piedade, M. E.; Martinho Simões, J. A. Coord. Chem.Rev. 1994 , 136/136, 737-807, and references cited therein.
(10) Minahan, D. M. A.; Hill, W. E.; McAuliffe, C. A. Coord. Chem. Rev. 1984 , 55,
(11) Levason, W. A.; McAuliffe, C. A. Adv. Inorg. Chem. Radiochem. 1972 , 14,
(12) Drent, E.; van Broekhoven, J. A. M.; Doyle, M. J. J. Organomet. Chem.1991 , 417, 235.
(13) Casey, C. P.; Whiteker, G. T. Isr. J. Chem. 1990 , 30, 299-304. (14) Steffen, W. L.; Palenik, G. Inorg. Chem. 1976 , 15, 2432-2439. (15) Hayashi, T.; Konishi, M.; Kumada, M. Tetrahedron Lett. 1979 , 21, 1871-
(16) Hayashi, T.; Konishi, M.; Kobori, Y.; Kumada, M.; Higuchi, T.; Hirotsu, K. J.Am. Chem. Soc. 1984 , 106, 158-163.
(17) Issleib, K.; Hohlfield, G. Z. Anorg. Allg. Chem. 1961 , 312, 169-179. (18) March, F. C.; Mason, R.; Thomas, K. M.; Shaw, B. L. J. Chem. Soc., Chem. Commun. 1975 , 584-585.
(19) Shaw, B. L. J. Organomet. Chem. 1975 , 94, 251-257. (20) Shaw, B. L. J. Organomet. Chem. 1980 , 200, 307-318, and references cited
(21) Al-Salem, N. A.; Empsall, H. D.; Markham, R.; Shaw, B. L.; Weeks, B. J.Chem. Soc., Dalton Trans. 1979 , 1972-1982.
(22) Hill, W. E.; Minahan, D. M. A.; Taylor, J. G.; McAuliffe, C. A. J. Am. Chem.Soc. 1982 , 104, 6001-6005.
(23) DeStefano, N. J.; Johnson, D. K.; Venanzi, L. M. Angew. Chem. 1974 , 86,
(24) DeStefano, N. J.; Johnson, D. K.; Venanzi, L. M. Helv. Chim. Acta 1976 , 59,
(25) Johnson, D. K.; Pregosin, P. S.; Venanzi, L. M. Helv. Chim. Acta 1976 , 59,
(26) Barrow, M.; Bürgi, H.-B.; Johnson, D. K.; Venanzi, L. M. J. Am. Chem. Soc.1976 , 98, 2356-2357.
(27) Reed, F. J. S.; Venanzi, L. M. Helv. Chim. Acta 1977 , 60, 2804-2814. (28) Bachechi, F.; Zambonelli, L.; Venanzi, L. M. Helv. Chim. Acta 1977 , 60,
(29) Baumgartner, E.; Reed, F. J. S.; Venanzi, L. M.; Bachechi, F.; Mura, P.;
Zambonelli, L. Helv. Chim. Acta 1983 , 66, 2572-2581.
(30) Holderegger, R.; Venanzi, L. M. Helv. Chim. Acta 1979 , 62, 2154-2158. (31) Camalli, M.; Caruso, F.; Chaloupka, S.; Venanzi, L. M. Helv. Chim. Acta 1988 , 71, 703-711.
(32) Bracher, G.; Grove, D. M.; Venanzi, L. M.; Bachechi, F.; Mura, P.;
Zambonelli, L. Helv. Chim. Acta 1980 , 63, 2519-2530.
(33) Baltensperger, U.; Günther, J. R.; Kägi, S.; Kahr, G.; Marty, W. Organometallics 1983 , 2, 571-578.
(34) Gillie, A.; Stille, J. K. J. Am. Chem. Soc. 1980 , 102, 4933-4941. (35) Aviron-Violet, P.; Colleuille, Y.; Varagnat, J. J. Mol. Catal. 1979 , 5, 41-50. (36) Casey, C. P.; Whiteker, G. T.; Campana, C. F.; Powell, D. R. Inorg. Chem. 1990 , 29, 3376-3381.
(37) Whiteker, G. T. Ph. D. Thesis, University of Wisconsin, 1990. (38) Kagan, H. B.; Dang, T. P. J. Am. Chem. Soc. 1972 , 6429-6433. (39) Casey, C. P.; Whiteker, G. T.; Melville, M. G.; Petrovich, L. M.; J.A. Gavney,
J.; Powell, D. R. J. Am. Chem. Soc. 1992 , 114, 5535-5543.
(40) Ball, R. G.; Trotter, J. Inorg. Chem. 1981 , 20, 261-265. (41) Hodgson, M.; Parker, D.; Taylor, R. J.; Ferguson, G. J. Chem. Soc., Chem. Commun. 1987 , 1309-1311.
(42) Casey, C. P.; Whiteker, G. T. J. Org. Chem. 1990 , 55, 1394-1396. (43) Yamamoto, K.; Momose, S.; Funahashi, M.; Miyazawa, M. Synlett 1990 ,
(44) Herrmann, W. A.; Kohlpaintner, C. W.; Herdtweck, E.; Kiprof, P. Inorg.Chem. 1991 , 30, 4271-4275.
(45) Sawamura, M.; Hashimoto, H.; Ito, Y. Tetrahedron Ass. 1991 , 2, 593. (46) Sawamura, M.; Hashimoto, H.; Ito, Y. Tetrahedron 1994 , 50, 4439-4454. (47) Sawamura, M.; Hashimoto, H.; Ito, Y. J. Am. Chem. Soc. 1992 , 114, 8295-
(48) Sawamura, M.; Kuwano, R.; Ito, Y. Angew. Chem., Int. Ed. Engl. 1994 , 33,
(49) Sawamura, M.; Hamashima, H.; Sugawara, M.; Kuwamo, R.; Ito, Y. Organometallics 1995 , 14, 4549-4558.
(50) Trost, B. M.; Breit, B.; Peukert, S.; Zambrano, J.; Ziller, J. W. Angew. Chem.,Int. Ed. Engl. 1995 , 34, 2386-2388.
(51) DBFphos has been synthesized previously: Haenel, M. W.; Jakubik, D.;
Rothenberger, E.; Schroth, G. Chem. Ber. 1991 , 124, 1705-1710.
PATIENT REGISTRATION Patient Information: Today’s Date:_____________ E-mail Address:_______________________________________________ Home Phone #__________________ Cel Phone #___________________Work Phone #___________________ Ext #______ Name______________________________________ I prefer to be cal ed_____________________Birthdate_______________ Address___________________________
ELENCO 2013 DELLE SOSTANZE PROIBITE DA NBFI, WNBF E INBF (dal 02/09/2009) La nostra associazione, a norma di legge, impone il rispetto delle regole dettate dal co-dice WADA. Le seguenti sostanze e preparati correlati sono vietati dalla NBFI, dalla INBFe dalla WNBF e il loro uso rappresenta motivo di espulsione dalle gare NBFI e la sospen-sione dei diritti di affiliazione all’organizzazion

 effects on the activity and selectivity induced by a catalyst. Forinstance Hayashi et al. showed that [dppf]PdCl2, a diphosphinewith a large P–Pd–P bite angle of 99.07°, induces very high activityand selectivity in the palladium-catalyzed cross-coupling of 2-butylmagnesiumchloride with bromobenzene, whereas application of[dppe]PdCl2 (with a P–Pd–P angle of 85.8°14) leads to no conversionat all.15,16
Casey et al. showed that in the rhodium-catalyzedhydroformylation the natural bite angle of the diphosphine ligandused has a dramatic influence on the regioselectivity of thereaction. BISBI (2,2’-bis((diphenylphosphino)methyl)-1,1’-biphenyl), a ligand that was shown to coordinate in a bisequatorialfashion (i.e. ∠P–Rh–P= ca. 120°) induces a ratio of linear tobranched aldehyde of up to 66 : 1, whereas dppe, whichcoordinates in a equatorial-axial fashion (i.e. ∠P–Rh–P= ca. 90°)leads to a linear to branched ratio of 2.1 : 1.8
Figure 2. Rhodium catalyzed hydroformylation
effects on the activity and selectivity induced by a catalyst. Forinstance Hayashi et al. showed that [dppf]PdCl2, a diphosphinewith a large P–Pd–P bite angle of 99.07°, induces very high activityand selectivity in the palladium-catalyzed cross-coupling of 2-butylmagnesiumchloride with bromobenzene, whereas application of[dppe]PdCl2 (with a P–Pd–P angle of 85.8°14) leads to no conversionat all.15,16
Casey et al. showed that in the rhodium-catalyzedhydroformylation the natural bite angle of the diphosphine ligandused has a dramatic influence on the regioselectivity of thereaction. BISBI (2,2’-bis((diphenylphosphino)methyl)-1,1’-biphenyl), a ligand that was shown to coordinate in a bisequatorialfashion (i.e. ∠P–Rh–P= ca. 120°) induces a ratio of linear tobranched aldehyde of up to 66 : 1, whereas dppe, whichcoordinates in a equatorial-axial fashion (i.e. ∠P–Rh–P= ca. 90°)leads to a linear to branched ratio of 2.1 : 1.8
Figure 2. Rhodium catalyzed hydroformylation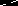 structure of Fe(CO)3(TBDCP ) reported later by Casey and Whiteker
structure of Fe(CO)3(TBDCP ) reported later by Casey and Whiteker


 high branched to linear ratios (97:3 for styrene; 98:2 for 2-methoxy-6-vinyl-naphtalene, a precursor for naproxen).43
BISBI, as already mentioned, supports large bite angles, but israther flexible. X-ray structural analyses of transition metalcomplexes of BISBI show a wide variety of P-M-P chelation angles:∠P-Mo-P= 103.5° in (BISBI)Mo(CO)4,44 ∠P-Ir-P= 117.9° in(BISBI)Ir(H)(CO)2, ∠P-Rh-P= 124.8° in (BISBI)Rh(H)(CO)(PPh3),39and ∠P-Fe-P= 152.0° in (BISBI)Fe(CO)3.36
A new group of ligands designed to span trans positions are thechiral (R,R)-(S,S)-2,2”-bis[1-(dialkylphosphino)ethyl]-1,1”-biferrocene (TRAP) ligands,45 which have been used effectively inthe rhodium-catalyzed asymmetric Michael reaction46,47 and therhodium-catalyzed hydrosilylation.48
X-ray crystal structures reveal large P-M-P chelation angles:∠P-Pd-P= 163.6° in (Ph-TRAP)PdBr2, and ∠P-Rh-P= 161.06° in(FurTRAP)Rh(CO)Cl.49TRAP is not a very rigid ligand; (small) quantities of cis-complexesare observed in the 31P NMR of crude reaction mixtures of theplatinum complexes.45 This flexibility is probably due to freerotation around the link between the aromatic rings in thebackbone, as was also found with BISBI.39
Very recently, Trost has introduced several new diphosphineligands which are able to support large bite angles for use in theasymmetric allylic alkylation.50 An X-ray crystal structure of a
high branched to linear ratios (97:3 for styrene; 98:2 for 2-methoxy-6-vinyl-naphtalene, a precursor for naproxen).43
BISBI, as already mentioned, supports large bite angles, but israther flexible. X-ray structural analyses of transition metalcomplexes of BISBI show a wide variety of P-M-P chelation angles:∠P-Mo-P= 103.5° in (BISBI)Mo(CO)4,44 ∠P-Ir-P= 117.9° in(BISBI)Ir(H)(CO)2, ∠P-Rh-P= 124.8° in (BISBI)Rh(H)(CO)(PPh3),39and ∠P-Fe-P= 152.0° in (BISBI)Fe(CO)3.36
A new group of ligands designed to span trans positions are thechiral (R,R)-(S,S)-2,2”-bis[1-(dialkylphosphino)ethyl]-1,1”-biferrocene (TRAP) ligands,45 which have been used effectively inthe rhodium-catalyzed asymmetric Michael reaction46,47 and therhodium-catalyzed hydrosilylation.48
X-ray crystal structures reveal large P-M-P chelation angles:∠P-Pd-P= 163.6° in (Ph-TRAP)PdBr2, and ∠P-Rh-P= 161.06° in(FurTRAP)Rh(CO)Cl.49TRAP is not a very rigid ligand; (small) quantities of cis-complexesare observed in the 31P NMR of crude reaction mixtures of theplatinum complexes.45 This flexibility is probably due to freerotation around the link between the aromatic rings in thebackbone, as was also found with BISBI.39
Very recently, Trost has introduced several new diphosphineligands which are able to support large bite angles for use in theasymmetric allylic alkylation.50 An X-ray crystal structure of a
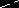
 Pd[allyl] complex of the most selective of these ligands (with ananthracene-based backbone) shows a P–Pd–P bite angle of 110.5°.
Pd[allyl] complex of the most selective of these ligands (with ananthracene-based backbone) shows a P–Pd–P bite angle of 110.5°.