Enantioselective Synthesis of -Aryl-γ-amino γ-amino acid derivatives have been reported in the past few
Acid Derivatives via Cu-Catalyzed Asymmetric
years,3 the search for new and efficient catalytic asymmetric
1,4-Reductions of γ-Phthalimido-Substituted
synthetic methods remains a significant challenge.
Very recently, we have reported a Rh-catalyzed asymmetric
, -Unsaturated Carboxylic Acid Esters
hydrogenation of γ-phthalimido-R, -unsaturated carboxylic acidesters, in which a variety of chiral -aryl-γ-amino acid deriva-
Jun Deng,†,‡ Xiang-Ping Hu,*,‡ Jia-Di Huang,†,‡
tives can be obtained with good enantioselectivities.4 However,
Sai-Bo Yu,†,‡ Dao-Yong Wang,†,‡ Zheng-Chao Duan,†,‡ and
this method has the disadvantages of demanding reaction
conditions (60 atm of H2 pressure), the use of the expensive
Dalian Institute of Chemical Physics, Chinese Academy of
Rh catalyst, and the relatively high catalyst loadings (1 mol
Sciences, Dalian 116023, China, and Graduate School of
%). These shortcomings prompted us to seek for an alternative
Chinese Academy of Sciences, Beijing 100039, China
approach to synthesize chiral -substituted γ-amino acids.
In the past decade, copper hydride (Cu-H) with chiral ligands
[email protected]; [email protected]
has emerged as a powerful reagent for effecting asymmetricreductions of various R, -unsaturated compounds5 such as
enones,6 R, -unsaturated esters,7 nitroalkenes,8 R, -unsaturatedsulfones,9 and R, -unsaturated nitriles.10 Since the copper saltsis very cheap in comparison with the Rh catalyst precursor, wetherefore envisioned that an asymmetric 1,4-reduction of γ-ph-thalimido-R, -unsaturated esters via copper hydride catalysisshould be an attractive alternative to these chiral compounds. Herein, we report our studies on this new strategy for construct-ing chiral -aryl substituted γ-amino acid derivatives.
We started our studies on the catalytic asymmetric conjugate
reduction of ethyl (Z)-4-phthalimido-3-phenylbut-2-enoate (1a) by surveying a number of copper salts, silanes, diphosphine ligands, and solvents in order to identify a suitable catalyst
-aryl-substituted γ-amino butyric acid
derivatives were synthesized in good enantioselectivities via
(2) (a) Olpe, H.-R.; Demieville, H.; Baltzer, V.; Bencze, W. L.; Koella, W. P.;
Wolf, P.; Haas, H. L. Eur. J. Pharmacol. 1978, 52, 133–136. (b) Sytinsky, I. A.;
the Cu-catalyzed asymmetric conjugate reduction of γ-ph-
Soldatenkov, A. T. Prog. Neurobiol. 1978, 10, 89–133. (c) Allan, R. D.; Bates,
thalimido-R, -unsaturated carboxylic acid esters using
M. C.; Drew, C. A.; Duke, R. K.; Hambley, T. W.; Johnston, G. A. R.; Mewett,
K. N.; Spence, I. Tetrahedron 1990, 46, 2511–2524. (d) Ong, J.; Kerr, D. I. S.;
2 H2O as a catalyst precursor, (S)-BINAP as a
Doolette, D. J.; Duke, R. K.; Mewett, K. N.; Allen, R. D.; Johnston, G. A. R.
ligand, PMHS as a hydride source, and t-BuOH as an
Eur. J. Pharmacol. 1993, 233, 169–172. (e) Costantino, G.; Macchiarulo, A.;
additive. The methodology has been applied successfully to
Guadix, A. E.; Pellicciari, R. J. Med. Chem. 2001, 44, 1827–1832. (f) Belliotti, T. R.; Capiris, T.; Ekhato, V.; Kinsora, J. J.; Field, M. J.; Hrffner, T. G.; Melzer,
the enantioselective synthesis of a chiral pharmaceutical,
L. T.; Schwarz, J. B.; Taylor, C. P.; Thorpe, A. J.; Vartanian, M. G.; Wise,
L. D.; Zhi-Su, T.; Weber, M. L.; Wustrow, D. J. J. Med. Chem. 2005, 48, 2294– 2307.
(3) For a review, see: Ordoˇnˇez, M.; Cativiela, C. Tetrahedron: Asymmetry2007, 18, 3–99.
(4) Deng, J.; Duan, Z.-C.; Huang, J.-D.; Hu, X.-P.; Wang, D.-Y.; Yu, S.-B.;
γ-Amino butyric acid (GABA) is an important central nervous
Xu, X.-F.; Zheng, Z. Org. Lett. 2007, 9, 4825–4828.
system neurotransmitter and has a profound impact on many
(5) For a review, see: Rendler, S.; Oestreich, M. Angew. Chem., Int. Ed.
important biological functions.1 Hence, many GABA analogues,
2007, 46, 498–504.
(6) (a) Moritani, Y.; Appella, D. H.; Jurkauskas, V.; Buchwald, S. L. J. Am.
particularly those bearing substituents at the -position such as
Chem. Soc. 2000, 122, 6797–6798. (b) Jurkauskas, V.; Buchwald, S. L. J. Am.
4-amino-3-(4-chlorophenyl)butyric acid (baclofen), have been
Chem. Soc. 2002, 124, 2892–2893. (c) Lipshutz, B. H.; Servesko, J. M. Angew.
well explored as medicines to treat various diseases associated
Chem., Int. Ed. 2003, 42, 4789–4792. (d) Lipshutz, B. H.; Servesko, J. M.; Petersen, T. B.; Papa, P. P.; Lover, A. A. Org. Lett. 2004, 6, 1273–1275. (e)
with GABA receptors.2 Studies have disclosed that the biological
Lipshutz, B. H.; Frieman, B. A.; Tomaso, Jr., A. E. Angew. Chem., Int. Ed 2006,
activities of these GABA analogues resides mainly in the single
(7) (a) Appella, D. H.; Moritani, Y.; Shintani, R.; Ferreira, E. M.; Buchwald,
enantiomer, and therefore the development of an enantioselective
S. L. J. Am. Chem. Soc. 1999, 121, 9473–9474. (b) Hughes, G.; Kimura, M.;
method for the synthesis of these compounds is highly desirable.
Buchwald, S. L. J. Am. Chem. Soc. 2003, 125, 11253–11258. (c) Lipshutz, B. H.;
Although some catalytic asymmetric syntheses of -substituted
Servesko, J. M.; Taft, B. R. J. Am. Chem. Soc. 2004, 126, 8352–8353. (d) Rainka, M. P.; Aye, Y.; Buchwald, S. L. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5821– 5823. (e) Rainka, M. P.; Milne, J. E.; Buchwald, S. L. Angew. Chem., Int. Ed.
† Dalian Institute of Chemical Physics. 2005, 44, 6177–6180. (f) Lipshutz, B. H.; Tanaka, N.; Taft, B. R.; Lee, C.-T.
‡ Graduate School of Chinese Academy of Sciences. Org. Lett. 2006, 8, 1963–1966.
(1) (a) Silverman, R. B.; Andruszkiewicz, R.; Nanavati, S. M.; Taylor, C. P.;
(8) (a) Czekelius, C.; Carreira, E. M. Angew. Chem., Int. Ed. 2003, 42, 4793–
Vartanian, M. G. J. Med. Chem. 1991, 34, 2295–2298. (b) Yuen, P. W.; Kanter,
4795. (b) Czekelius, C.; Carreira, E. M. Org. Lett. 2004, 6, 4575–4577. (c)
G. D.; Taylor, C. P.; Vertanian, M. G. Bioorg. Med. Chem. Lett. 1994, 4, 823–
Czekelius, C.; Carreira, E. M. Org. Process Res. DeV. 2007, 11, 633–636.
826. (c) Bryans, J. S.; Davies, N.; Gee, N. S.; Dissanayake, V. U. K.; Ratcliffe,
(9) Llamas, T.; Arraya´s, R. G.; Carretero, J. C. Angew. Chem., Int. Ed. 2007,
G. S J. Med. Chem. 1998, 41, 1838–1845. (d) Moglioni, A. G.; Brousse, B. N.;
´ lvarez-Larena, A.; Moltrasio, G. Y.; Ortun˜o, R. M. Tetrahedron: Asymmetry
(10) (a) Lee, D.; Kim, D.; Yun, J. Angew. Chem., Int. Ed. 2006, 45, 2785– 2002, 13, 451–454.
2787. (b) Lee, D.; Yang, Y.; Yun, J. Org. Lett. 2007, 9, 2749–2751. J. Org. Chem. 2008, 73, 6022–6024
10.1021/jo800794p CCC: $40.75 2008 American Chemical Society
Enantioselective Conjugate Reduction of (Z)-4-Phthalimido-3-phenylbut-2-enoate (1a)a a All reactions were conducted using 0.25 mmol of substrate, 5 mol % Cu, 5 mol % ligand, 1.0 mmol of t-BuOH, and 1.0 mmol of silane in 2 mL of
solvent at room temperature for 24 h, unless otherwise specified. b Isolated yields. c Values were determined by HPLC on a chiral column (ChiralpakAD or AD-H). d Absolute configuration was determined by comparison of the sign of the optical rotation with the reported data. e No added t-BuOH. f Not determined because of low reactivity.
system. The results are summarized in Table 1. Initially, weemployed the published procedure7a for the preparation of thecatalyst formed between CuCl, BINAP, and NaOt-Bu. To ourdelight, this catalyst promoted the reaction in good enantiose-lectivity and moderate yield by using PMHS as the stoichio-metric hydride donor (entry 1). Other copper salts were thenscreened
2 H2O12 is the best catalyst precursor in terms of yield
and enantioselectivity, providing the reduction product in 78%yield and 96% ee (entry 4). Since BINAP has proved to be anefficient ligand for this transformation, some structurally similardiphosphines were next investigated (Figure 1). Although good
FIGURE 1. Representative ligands for asymmetric 1,4-reduction.
enantioselectivities were obtained in some cases, no resultssurpassed that obtained with BINAP (entries 4-7). Subsequent
experiments in an effort to increase yield and enantioselectivity
2 H2O as the catalyst precursor, BINAP as the ligand,
by the variation of the silane reagents proved unsuccessful. We
PMHS as the silane reagent, and toluene as the solvent for
found that the use of TMDS, diphenylsilane or phenylsilane in
investigating the scope of this new method on various ethyl
the reaction resulted in dramatically decreased reaction rates
(Z)-4-phthalimido-3-arylbut-2-enoate (1), and the results are
(entries 8-10). Solvent-screening experiments revealed that the
summarized in Table 2. Initially, a variety of substituted (Z)-
nature of the solvents had a profound effect on the catalytic
4-phthalimido-3-phenylbut-2-enoates (1b-h) were examined,
reaction. The reactions performed in THF gave an increased
and the results indicated that the electronic properties of the
enantioselectivity, but decreased reaction rate (entry 11). Using
substituent in the phenyl ring had little effect on the enantiose-
xylene as the solvent, a comparable result was achieved (entry
lectivity. All of the substrates were reduced in 93-96% ee
12). However, very low reaction rate was observed when the
(entries 1-7). These reduction products can be easily upgraded
reaction was carried out in CH2Cl2 (entry 13).
via recrystallization to higher level of ee values because of theirhigh crystallinity conferred by the phthalimido group. Good
(11) For copper fluorides as useful catalyst precursors in the Cu-H catalyzed
enantioselectivities were also obtained in the conjugate reduction
hydrosilylation, see: Sirol, S.; Courmarcel, J.; Mostefai, N.; Riant, O. Org. Lett. 2001, 3, 4111–4113.
of -2-naphthyl- and -2-thiophenyl-substituted substrates (1i-j,
(12) For copper(II) acetate or copper(II) acetate monohydrate as useful
entries 8 and 9). These results demonstrated the applicability
catalyst precursor in the Cu-H catalyzed hydrosilylation, see: Lee, D.; Yun, J.
of this new method in the enantioselective synthesis of
Tetrahedron Lett. 2004
(13) (a) Anada, M.; Hashimoto, S. Tetrahedron Lett. 1998, 39, 79–82. (b)
Resende, P.; Almeida, W. P.; Coelho, F. Tetrahedron: Asymmetry 1999, 10,
To explore the potential synthetic utility of this new method,
2113–2118. (c) Corey, E. J.; Zhang, F.-Y. Org. Lett. 2000, 2, 4257–4259. (d) Thakur, V. V.; Nikalje, M. D.; Sudalai, A. Tetrahedron: Asymmetry 2003, 14,
we attempted its application in the synthesis of the chiral
581–586. (e) Meyer, O.; Becht, J.-M.; Helmchen, G. Synlett 2003, 1539–1541.
pharmaceuticals (R)-baclofen (Scheme 1). Although baclofen
(f) Becht, J.-M.; Meyer, O.; Helmchen, G. Synthesis 2003, 2805–2810. (g) Belda, O.; Lundgren, S.; Moberg, C. Org. Lett. 2003, 5, 2275–2278. (h) Felluga, F.;
is commercialized in its racemic form, pharmacological studies
Gombac, V.; Pitacco, G.; Valentin, E. Tetrahedron: Asymmetry 2005, 16, 1341–
have shown that its biological activity resides exclusively in its
1345. (i) Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. J. Am.
(R)-enantiomer.2a As we have reported, the requisite substrate
Chem. Soc. 2005, 127, 119–125. (j) Paraskar, A. S.; Sudalai, A. Tetrahedron 2006, 62, 4907–4916.
(Z)-3-(4-chlorophenyl)-4-phthalimidobut-2-enoate (1c) was pre- J. Org. Chem. Vol. 73, No. 15, 2008Enantioselective Conjugate Reduction of γ-phthalimido groups, which has led to the asymmetric synthesis
(Z)-4-Phthalimido-3-arylbut-2-enoate (1) with a Cu(OAc) ·
of various -aryl-substituted γ-amino acid derivatives in good
(S)-BINAP/PMHS Catalytic Systema
enantioselectivities. Interestingly, the results disclosed that theheteroatom in the γ-position seems to be no impact on thedirection of hydride delivery. High crystallinity conferred bythe phthalimido group made the upgrade of ee values of thereduction products possible. This method has been successfullyapplied in the enantioselective synthesis of the chiral pharma-
1b: Ar ) 4-FC6H4 Experimental Section 1c: Ar ) 4-ClC6H4
Ethyl (Z)-4-phthalimido-3-arylbut-2-enoates 1a-j were prepared 1d: Ar ) 4-BrC6H4 1e: Ar ) 4-MeOC6H4 General Procedure for Catalytic Asymmetric 1,4-Reduction. 1f: Ar ) 4-CF3C6H4 1g: Ar ) 3-MeOC
2 H2O (5 mg, 0.025 mmol), (S)-BINAP (15.6 mg, 0.025
1h: Ar ) 3-cyclopentoxy-4-MeOC6H3
mmol), and toluene (1.0 mL) were added into an oven-dried Schlenk
1i: Ar ) 2-naphthyl
tube. The resulting mixture was stirred at room temperature for 30
1j: Ar ) 2-thiophenyl
min. Then, PMHS (0.12 mL, 2.0 mmol) was added to the reaction
mixture, which was stirred for 30 min. A solution of ethyl (Z)-3-
Reactions were carried out with 0.5 mmol of substrate in 2 mL of
(4-chlorophenyl)-4-phthalimidobut-2-enoate (1c) (185 mg, 0.5
toluene at room temperature for 24 h, with a substrate/Cu(OAc)2 H2O/
mmol) in toluene (1.0 mL) was added, followed by t-BuOH (0.191
(S)-BINAP/PMHS/t-BuOH ratio of 1/0.05/0.05/4/4. b Isolated yields. c Values were determined by HPLC on a chiral column (Chiralpak
mL, 2.0 mmol). The reaction vessel was sealed, and the mixture
AD-H or chiralcel OD-H). d Absolute configuration was determined by
was stirred for 24 h. The reaction mixture was quenched with
comparison of the sign of the optical rotation with the reported data.
saturated aqueous ammonium chloride solution, and the aqueouslayer was extracted with Et2O (3 × 20 mL). The combined organic
SCHEME 1. Synthesis of (R)-Baclofen
layers were washed with brine, dried over MgSO4, and concentrated. The product was purified by chromatography on silica gel. Ethyl (S)-3-(4-Chlorophenyl)-4-phthalimidobutanoate (2c). 1H
NMR (400 MHz, CDCl3): δ 1.08 (t, J ) 7.2 Hz, 3H), 2.68-2.72(m, 2H), 3.72-3.75 (m, 1H), 3.86-3.91 (m, 2H), 3.92 (q, J ) 7.2Hz, 2H), 7.20-7.26 (m, 4H), 7.69-7.71 (m, 2H), 7.79-7.81 (m,2H). 13C NMR (100 MHz, CDCl3): δ 14.1, 38.6, 40.2, 42.9, 60.6,123.3, 128.8, 129.1, 131.8, 133.0, 134.1, 138.9, 168.1, 171.2. HRMS(ESI) m/z calcd for C20H18NO4ClNa+ 394.0822, found 394.0836. [R]25 -
60.4 (c 0.5, CHCl3). 93% ee was determined by chiral
HPLC (Chiralpak AD-H (0.46 cm × 25 cm), i-PrOH/hexane )15/85, UV 254 nm, 40 °C, 1.0 mL/min), retention times (min) 16.4(major, S) and 22.2 (minor, R). Synthesis of (R)-Baclofen. 13 A mixture of (R)-2c (186 mg, 0.5
mmol, 94% ee) and 6 M HCl (10 mL) was heated under reflux for12 h. The solution was cooled, and the precipitated phthalic acidwas filtered off. The filtrate was evaporated to dryness, and theresulting solid was resuspended in water (10 mL). The filtrate wasevaporated to dryness under reduced pressure and then dried undervacuum to give 112 mg (90.2% yield) of (R)-baclofen as a colorlesssolid, mp 198-199 °C; [R]25 -
pared through a three-step transformation from 4′-chloroac-
2O). 1H NMR (400 MHz, D2O): δ 2.73-2.90 (m, 2H),
3.24-3.47 (m, 3H), 7.35-7.48 (m, 4H). 13C NMR (100 MHz,
etophenone in good yields (over 60% in total yields). With the
D2O): δ 38.3, 39.4, 43.7, 129.3, 129.5, 133.4, 137.1, 175.3.
catalysis of Cu/(R)-BINAP, 1c was reduced in 92% yield and 94% ee. The resulting reduction product 2c was further upgraded Acknowledgment. We are grateful for financial support from
via recrystallization to over 98% ee and then converted in one
the National Natural Science Foundation of China (20472083).
step to (R)-baclofen in a nearly quantitative yield, demonstrating
Supporting Information Available: Experimental details,
the potential utility of this method in the synthesis of chiral
and analysis of ee-values of products. This material is available
free of charge via the Internet at http://pubs.acs.org.
In conclusion, we have developed a method for the asym-
metric conjugate reduction of R, -unsaturated esters containing
J. Org. Chem. Vol. 73, No. 15, 2008
Medical Treatment __________________________________________ Not all children with congenital heart defects require treatment. Some may only need to be observed and visit their cardiologist. In other case, surgery or cardiac catheterization may be needed to reduce the effects of and/or repair the defect. Whether babies need surgical or catheterization treatment, they can develop conditions
MODULE 13 Growing the section workbook Appendices scouts.org.uk/appointment APPENDIX 1: DISCUSSION POINT 2 – VOLUNTEERING RESPONSES Ask them! Don’t assume that people know you need help or that they would be comfortable offering it. Talk to those around you and get to know parents. Once you find out more about them, you may be able to ask them to do specific tasks or ac
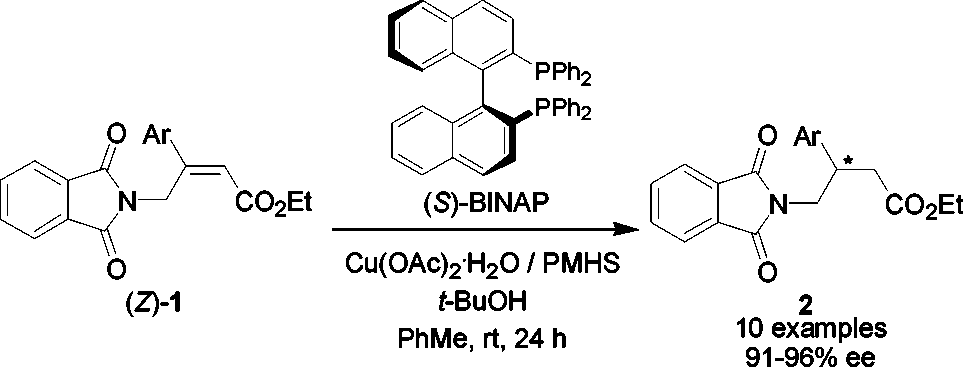 Enantioselective Synthesis of -Aryl-γ-amino
Enantioselective Synthesis of -Aryl-γ-amino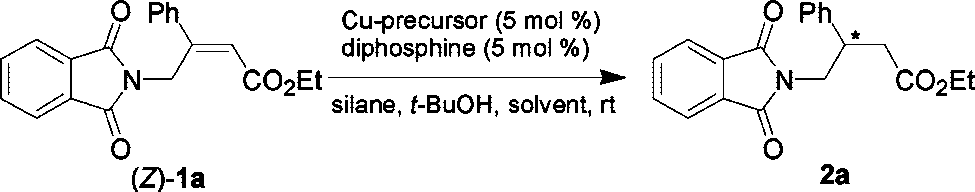
 Enantioselective Conjugate Reduction of (Z)-4-Phthalimido-3-phenylbut-2-enoate (1a)a
Enantioselective Conjugate Reduction of (Z)-4-Phthalimido-3-phenylbut-2-enoate (1a)a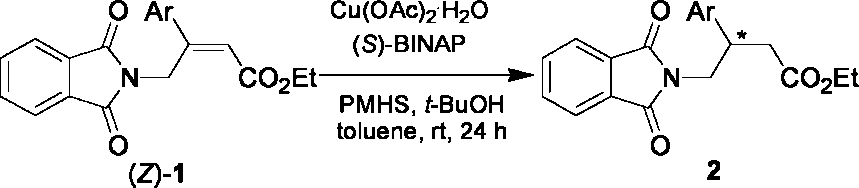
 Enantioselective Conjugate Reduction of
Enantioselective Conjugate Reduction of